Publications
*Contributed equally
†Corresponding author
[29] Deenalattha DHS, Jurich CP, Lange B, Armstrong D, Nein K, Yesselman JD†, “Characterizing RNA 3D structural features from DMS reactivity” (2025), Nucleic Acid Research, In revision (Preprint)
[28] Cornwell-Arquitt RL, Nigh R, Hathaway MT, Yesselman JD†, Hendrix DA†, “Analysis of natural structures and chemical mapping data reveals local stability compensation in RNA”(2025), Nucleic Acid Research, Volume 53, Issue 12, Pages gkaf565 (Link)
[27] Lange B, Gil RG, Yesselman JD†, “High-throughput determination of RNA tertiary contact thermodynamics by quantitative DMS chemical mapping”(2024), Nucleic Acid Research, Volume 52, Issue 16, Pages 9953–9965 (Link)
[26] Bu F, Miao Z, Ali Ibrahim Adam Y, Adamiak RW, Antczak M, Rebeca H. de Aquino B, Badepally NG, Batey RT, Baulin EF, Boinski P, Boniecki MJ, Bujnicki JM, Carpenter KA, Chacon J, Chen S, Chiu W, Cordero P, Das NK, Das R, Dawson W, Dimaio F, Ding F, Dock-Bregeon AC, Dokholyan NV, Dror R, Dunin-Horkawicz S, Eismann S, Ennifar E, Esmaeeli R, Farsani MA, Ferré-D’Amaré AR, Geniesse C, Guzman HV, Hood IV, Huang L, Jain DS, Jaryani F, Jin L, Joshi A, Karelina M, Kieft JS, Kladwang W, Kmiecik S, Koirala D, Kretsch R, Kurcinski M, Lee S, Li S, Li J, Magnus M, Masquida B, Moafinejad SN, Mondal A, Mukherjee S, Nguyen THD, Nikolaev G, Nithin C, Nye G, Perez A, Pham P, Piccirilli JA, Pilla SP, Nayaka IP, Pluta R, Poblete S, Ponce-Salvatierra A, Popenda L, Popenda M, Pucci F, Rangan R, Ray A, Ren A, Sanbonmatsu K, Sarzynska J, Sha CM, Stefaniak F, Su Z, Suddala KC, Szachniuk M, Townshend R, Trachman RJ, Wang W, Wang J, Watkins A, Wirecki T, Xiao Y, Xiong Y, Xiong P, Yang J, Yesselman JD, Zhang J, Zhang S, Zhang D, Zhang Y, Zhang Z, Zhou Y, Zok T, Żyla A, Westhof E†, RNA-Puzzles Round V: The blind predictions of 23 RNA structures (2025), Nature Methods, 22, pages 399–411 (Link)
[25] Jurich C, Jeppesen M, Sakallioglu I, De Lima Leite A, Yesselman JD†, Powers B†, A simulated LC-MS dataset for assessing the metabolomics data processing pipeline implemented into MVAPACK (2024), Analytical Chemistry, 96, 32, 12943–12956 (Link)
[24] Camara MB, Lange B, Yesselman JD, Eichhorn CD†. “Visualizing a two-state conformational ensemble in stem–loop 3 of the transcriptional regulator 7SK RNA” (2024), Nucleic Acids Research, Volume 52, Issue 2 (Link)
[23] Jurich CP, Brivanlou A, Rouskin S†, Yesselman JD† (2022), “Web-based platform for analysis of RNA folding from high throughput chemical probing data”, Nucleic Acids Research, Volume 50, Issue W1 (Link)
[22] Van Damme R., Li K, Zhang M, Bai J, Wilson LH, Yesselman JD, Zhipeng L†, Velema WA† (2022) Chemical reversible crosslinking enables measurement of RNA 3D distances and alternative conformations in cells. Nature Communications 13, 911. (Link)
[21] Zafferani M, Haddad C, Davila-Calderon J, Yuan-Chiu L, Mugisha CS, Monaghan AG, Kennedy AA, Yesselman JD, Gifford RR, Tai AW, Kutluay SB, Li ML, Brewer G, Tolbert BS, Hargrove AE† (2020) “Amilorides inhibit SARS-CoV-2 replication in vitro by targeting RNA structures”, Science Advances 7, 48. (Link)
[20] Jurich CP, Yesselman JD † , (2020) “Automated 3D design and evaluation of RNA nanostructures with RNAMake”, Methods in Molecular Biology (Link)
[19] Shi H, Rangadurai A, Assi HA, Roy R, Case DA†,, Herschlag D†, Yesselman JD † , Al- Hashimi HM†, (2020) “Rapid and Accurate Determination of Atomistic RNA Dynamic Ensemble Models Using NMR and Structure Prediction”, Nature Communications 11, 5531, PMID: 33139729 (Link)
[18] Miao Z, Adamiak RW, Antczak M, Boniecki MJ, Bujnicki JM, Chen SJ, Cheng CY, Cheng Y, Chou FC, Das R, Dokholyan NV, Ding F, Geniesse C, Jiang Y, Joshi A, Krokhotin A, Magnus M, Mailhot O, Major F, Mann TH, Piatkowski P, Pluta R, Popenda M, Sarzynska J, Sun L, Szachniuk M, Tian S, Wang J, Wang J, Watkins AM, Wiedemann J, Xiao Y, Xu X, Yesselman JD, Zhang D, Zhang Y, Zhang Z, Zhao C, Zhao P, Zhou Y, Zok T, Zyla A, Ren A, Batey RT, Golden BL, Huang L, Lilley DM, Liu Y, Patel DJ, Westhof E (2020). “RNA- Puzzles Round IV: 3D structure predictions of four ribozymes and two aptamers”, RNA, 26: 982-995. (Link)
[17] *Kappel K, *Zhang K, Su Z, Kladwang W, Li S, Pintilie G, Topkar VV, Rangan R, Zheludev IN, Watkins AM, Yesselman JD, Chiu W, Das R “Accelerated cryo-EM-guided determination of three-dimensional RNA-only structures”, Nature Methods, 17, pgs 699–707 (Link)

Ribosolve is a computational / experimental pipeline to determine RNA-only structures.
Functional insights from Ribosolve models. (A) Overlay of V. cholerae and F. nucleatum glycine riboswitches with glycine. (B) Overlay of both glycine aptamers from the F. nucleatum and V. cholerae structures. (C-D) Structural homology between the SAM-IV riboswitch and SAM-I and SAM-I/IV riboswitches. (C) The SAM-I crystal structure (37), the SAM-I/IV crystal structure (36), and the SAM-IV Ribosolve model, and (D) an overlay of all three structures, with peripheral elements shown as gray transparent cartoons. SAM is shown as transparent red spheres. (E) The computationally designed eterna3D-JR_1 structure (gray) overlaid with the Ribosolve model (purple). (E-G) Comparisons of Ribosolve structures and the computationally designed models for (E) Eterna3D-JR_1, (F) ATP-TTR-3 with and without AMP, and (G) Spinach-TTR-3.
[16] *Yesselman JD, *Denny SK, Bisaria N, Herschlag D, Greenleaf WJ, Das R (2019) “Sequence-dependent RNA helix conformational preferences predictably impact tertiary structure formation”, in press, epub available, Proceedings of the National Academy of Sciences U.S.A (Link | Paper | Download Software)

RNAMake-ΔΔG accounts for changes in tertiary RNA assembly affinity in a blind prediction challenge.
Scatterplot compares the dependence of the observed changes in ΔΔG (compared to the median) on the RNAMake-ΔΔG model for 1536 chip piece variants (R = 0.84). Red dashed line indicates the best-fit line (slope = 0.54); cyan dotted line indicates the line of slope 1.
[15] Yesselman JD, Eiler D, Carlson ED, Gotrik MR, d'Aquino AE, Ooms AN, Kladwang W, Shi X, Costantino D, Lucks JB, Herschlag D, Jewett MC, Kieft JS, Das R "Computational Design of Asymmetric Three-dimensional RNA Structures and Function” Nature Nanotechnology, 14, 866–873 (2019).(Link | Paper | Download Software)

Problems in RNA nanotechnology solved by RNAMake
(a) ‘miniTTRs’ require two strands (green, purple between tetraloop (orange) and tetraloop-receptor (blue); (b) tethered ribosomes require two strands (green, purple) to link the small subunit (orange) to the large subunit (blue). c) ‘Locking’ a small-molecule binding aptamer (cyan; ATP molecule in pink spheres) by designing four strands (green, purple, teal, magenta) to a peripheral tertiary contact(orange, blue). d) Demonstration of RNAMake design algorithm, which builds an RNA path via the successive addition of motifs and helices from a starting base pair to the ending base pair.

High throughput measurements of RNA tertiary structure energetics
Characterizing the thermodynamic fingerprints of >1,000 RNA junctions reveals principles for how RNA sequence affects tertiary assembly energetics, highlighting a path toward tertiary folding prediction by integrating static structural and dynamic energetic information.

RMDB simplifies chemical mapping data distribution
Screenshot of the new interactive user interface for viewing RMDB entries: An example of an entry.

M2-seq recovers helices across diverse RNA folds
M2-seq recovers the secondary structure of the P4–P6 domain of Tetrahymena ribozyme. Depicts the crystallographic secondary structure and M2-seq data (square graphs) with colored labels (on both display items) marking helices and multihelix domains automatically identified by M2-net analysis (A neural network).
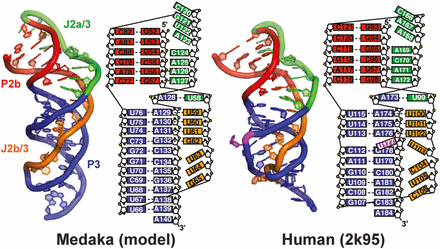
Structural comparison between medaka and human telemerase pseudoknot
Comparison of minimal mdPK and hPK (PDB ID code 2K95) structures. Secondary structure elements are P2b (red), P3 (blue), J2a/3 (green), and J2b/3 (gold).

Model of active state transition in Tetrahymena ribozyme
Models for active site interactions within (E•S•G)O and (E•S•G)C.. The black arrows highlight changes in the positions of active site residues in going from (E•S•G)C to (E•S•G)O.

FARFAR RNA 3D prediction accuracy
A) GCAA tetraloop (1ZIH), RNA Denovo lowest energy models displays a high level of convergence. B) Pseudoknot (1L2X), less converged then tetraloop but also larger, still within 3Å heavy-atom rmsd for top model. C) 4x4 internal loop solved by NMR at PDB ID 2L8F, converges despite presenting 4 non-canonical base pairs.

Accessing accurate RNA 3D Rosetta modeling
RP domain IV RNA (PDB ID: 1LNT) contains highly conserved AC base pairs that RNA-Redesign mutates to stabilize the RNA

Schematic and runtime of the primerize algorithm
Schematic of the Primerize algorithm. Tm (STEP 1) and misprime matrices (STEP 2) are pre-calculated for the dynamic programming assembly.

Survey of C---OH hydrogen bonds in proteins
Depiction of angles and distances measured. B: Methyl hydrogen donor to acceptor distances in which the acceptor is oxygen (solid line) or carbon (dashed line). Dashed-dot line is the difference of the latter curves. C: Elevation angles of methyl CH···O hydrogen bonds. D: Methyl CH···X angles in which X is oxygen (solid line) or carbon (dashed line).
[5] Horowitz S, Dirk LM, Yesselman JD, Nimtz JS, Adhikari U, Mehl RA, Scheiner S, Houtz RL, Al-Hashimi HM, Trievel RC (2013) "Conservation and functional importance of carbon-oxygen hydrogen bonding in AdoMet-dependent methyltransferases" Journal of the American Chemical Society 16;135(41):15536-48 (Link | Paper)

Six classes of adomet-dependent methyltransferases
The hydrogen-bond donor and methyl C···O interaction distances are labeled in each enzyme.

Chemical classes requiring additional refinement
Average unsigned errors of hydration free energies for specific chemical classes for (top panel) CGENFF molecules and (bottom panel) non-CGENFF compounds.

Quality of the minimized MATCH-typed molecules
PubChem drug-like molecules that were successfully processed using the CGENFF libraries within MATCH to generate their respective topology and parameter files. RMSD was computed by comparing conformations found in the PubChem database to the ones after minimization.

Locations of ionizable residues in Δ+PHS
Δ+PHS staphylococcal nuclease is shown here with all ionizing residues highlighted. Glutamic acid is cyan, and aspartic acid is orange. (From: Predicting extreme pKa shifts in staphylococcal nuclease mutants with constant pH molecular dynamics

Optimized active site with bound adomet
Truncated AdoMet and the protein are depicted with green and gray carbon atoms, respectively. Residues labeled in red designate CH O acceptors. H O distances from methyl protons to nearest oxygen atom for optimized and broken geometry are shown in magenta and cyan, respectively.